Atoms are the building blocks of matter and the interactions between them shape the properties of molecules and materials. Understanding these interactions through molecular dynamics simulations is crucial for various scientific fields such as drug development and material design. However, the computational cost of simulating these interactions using traditional methods has been a significant challenge.
The Schrödinger equation, which describes the energy levels of quantum systems like atoms and molecules, is the foundation of traditional methods for computing electronic interactions. However, solving this equation for molecules with a large number of atoms can be time-consuming, even on powerful computers. This has limited the ability to run molecular dynamics simulations over long time-scales, hindering progress in research areas like protein folding and material design.
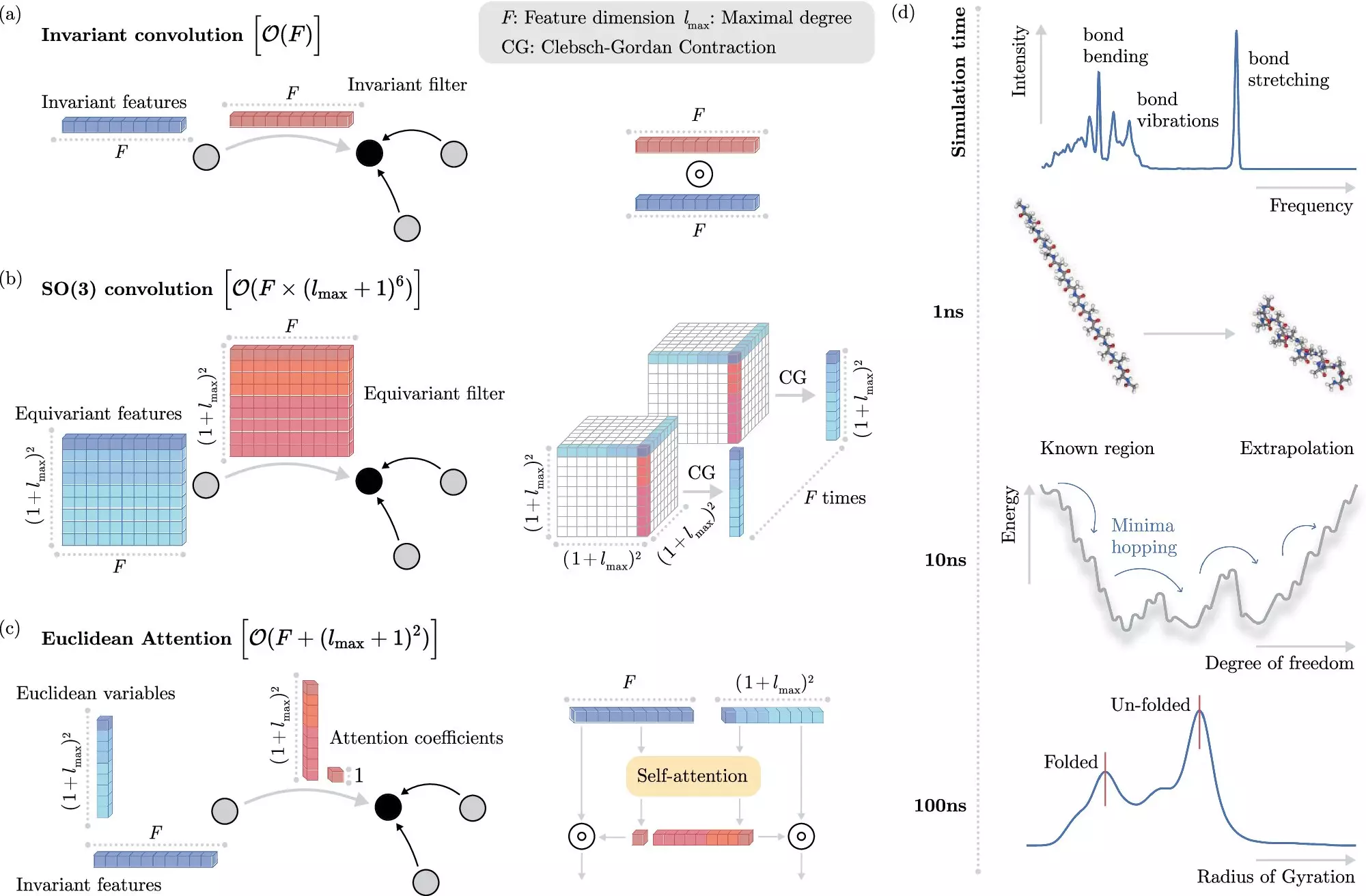
In recent years, machine learning (ML) has emerged as a powerful tool to overcome the challenges posed by traditional methods. By directly predicting electronic interactions at the atomistic level, ML algorithms have significantly reduced the computational cost of simulating molecular dynamics. However, incorporating invariances into ML models has been a computationally expensive process, limiting the speed at which these models can operate.
Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind have developed a novel machine learning algorithm that addresses this limitation. By decoupling invariances from other information about a chemical system at the outset, the new approach simplifies the process and allows the ML model to focus on the most critical operations. This breakthrough has enabled simulations that previously required months or even years to be performed within a few days on a single computer node.
The increased efficiency of molecular dynamics simulations opens up new possibilities for research and development. The accurate simulation of molecules interacting with proteins in the human body, for example, could revolutionize drug development by eliminating the need for costly and time-consuming experiments. Additionally, the ability to quickly identify stable molecular configurations, as demonstrated with docosahexaenoic acid, can advance our understanding of complex biological systems.
As noted by Prof. Dr. Klaus-Robert Müller, the combination of advanced machine learning techniques with physical principles represents a critical advancement in computational chemistry. The next generation of algorithms will need to accurately simulate complex system sizes and interactions, paving the way for a deeper understanding of the fundamental processes of nature.
The development of a new machine learning algorithm for molecular dynamics simulations represents a significant breakthrough in the field of computational chemistry. By overcoming the computational challenges associated with traditional methods, this innovation has the potential to revolutionize scientific research and development in areas such as drug discovery and material design. The future of molecular dynamics simulations is bright, thanks to the combination of advanced machine learning techniques and physical principles.
Leave a Reply